DISCO: Designing Sequence
and Structure as One
DISCO, DIffusion for Sequence-structure CO-design, is a multimodal generative model that simultaneously generates both a protein's amino acid sequence and its three-dimensional atomic structure. This might sound like an incremental advance, but it represents a fundamental departure from how the field has worked.
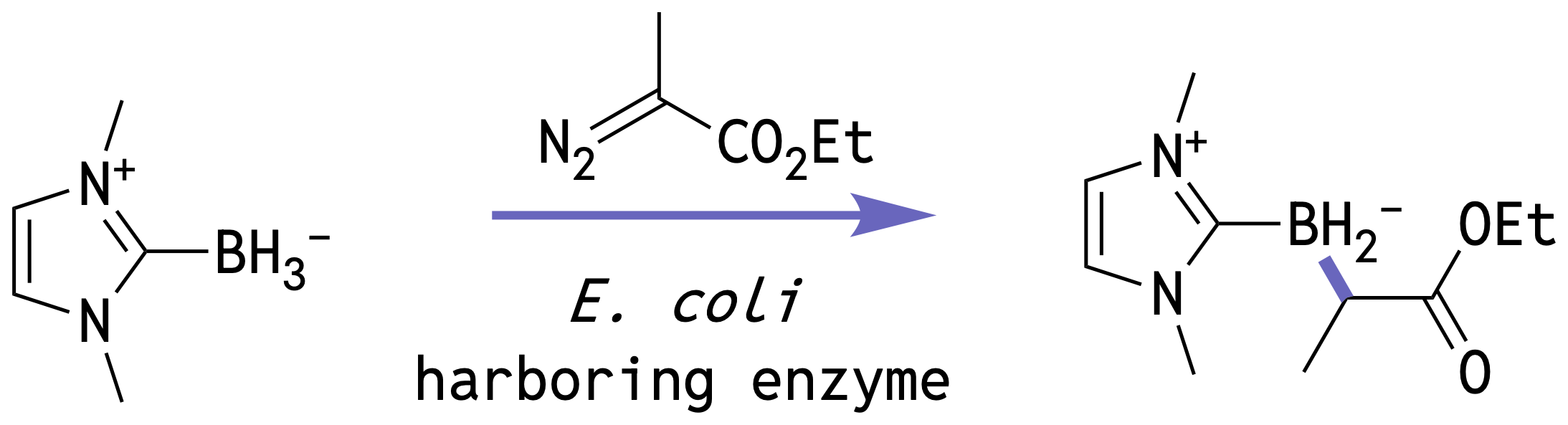
The wet-lab pipeline of existing generative approaches, including recent models like RFdiffusion2[5,6], RFdiffusion3[7], and BoltzGen[8], work in two separate stages: first generate a protein backbone, then use a separate inverse-folding network such as LigandMPNN[9] to predict what amino acid sequence would fold into that shape. This decoupled pipeline cannot use sequence-level signals to guide backbone design, or structural context to inform sequence choices.
DISCO eliminates this handoff entirely. It learns a joint distribution over discrete amino acid tokens and continuous 3D coordinates, denoising both simultaneously. The mathematical foundation is elegant: by independently sampling noise per modality during training, the model provably learns the joint reverse process using only unimodal losses, no special joint supervision required[10]. The coupling emerges from the architecture.
Core Techniques
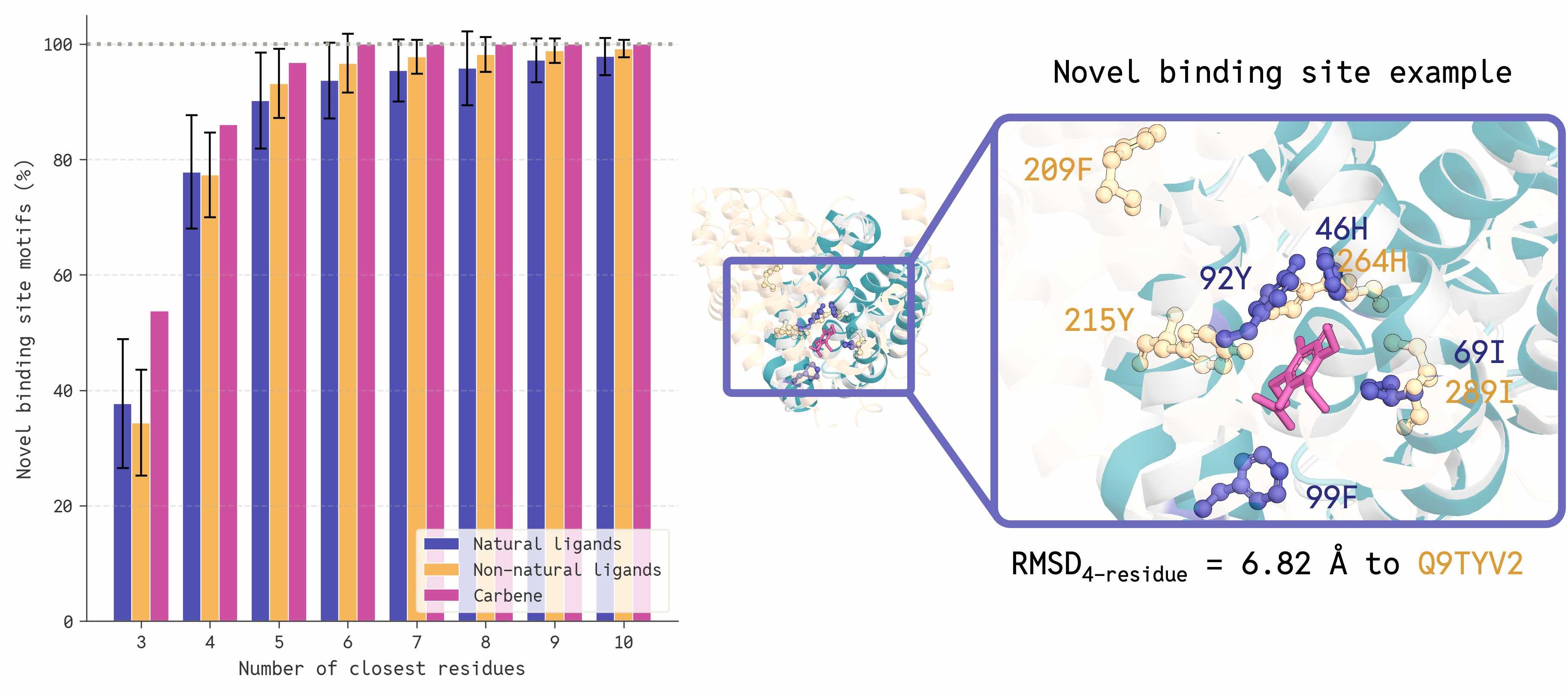
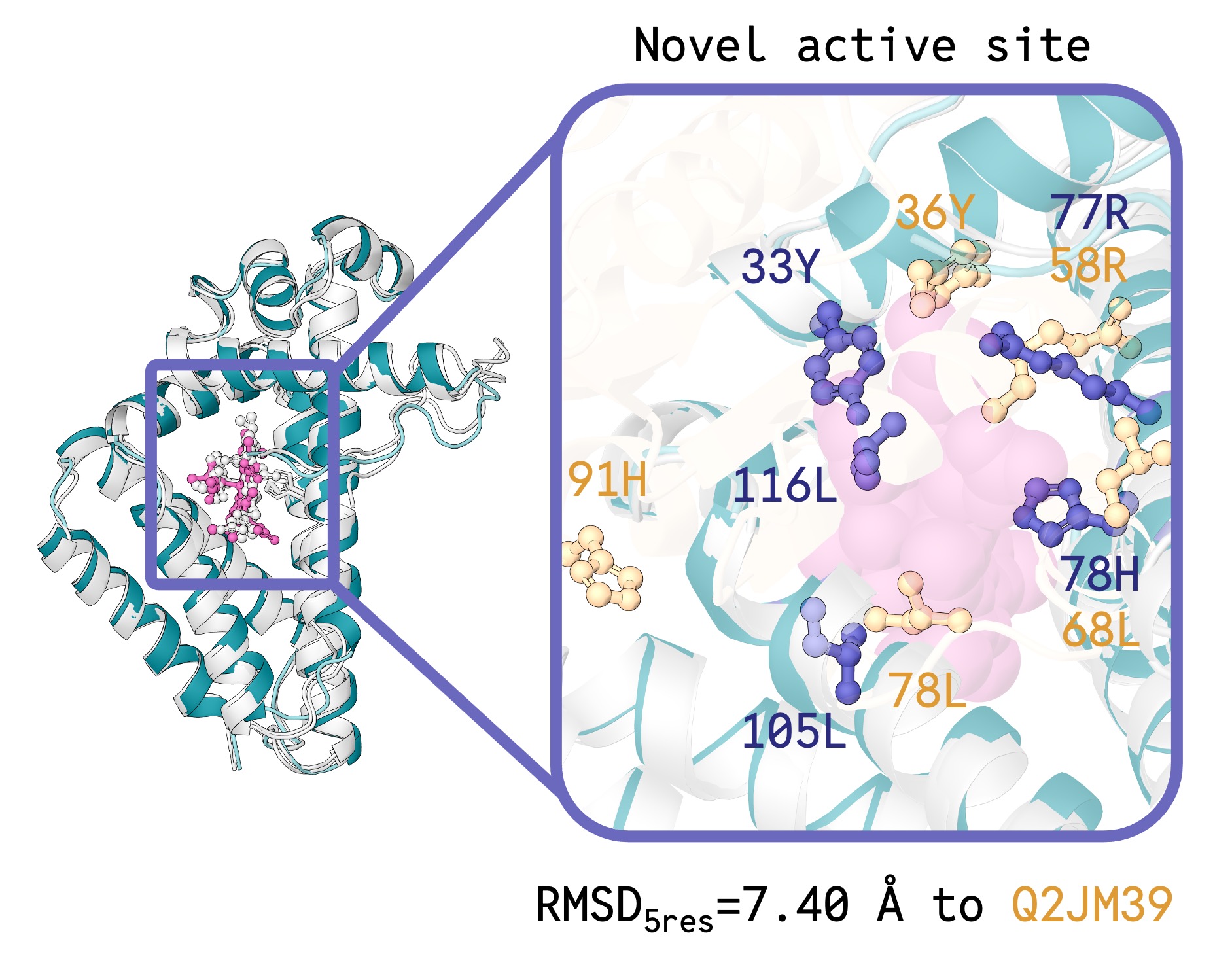
Getting the model to generate sequences that actually fold into their designed structures requires us to align the two modalities together, which includes three critical inference innovations, each with a dramatic effect on co-designability:
Some other highlights of the model are as follows:
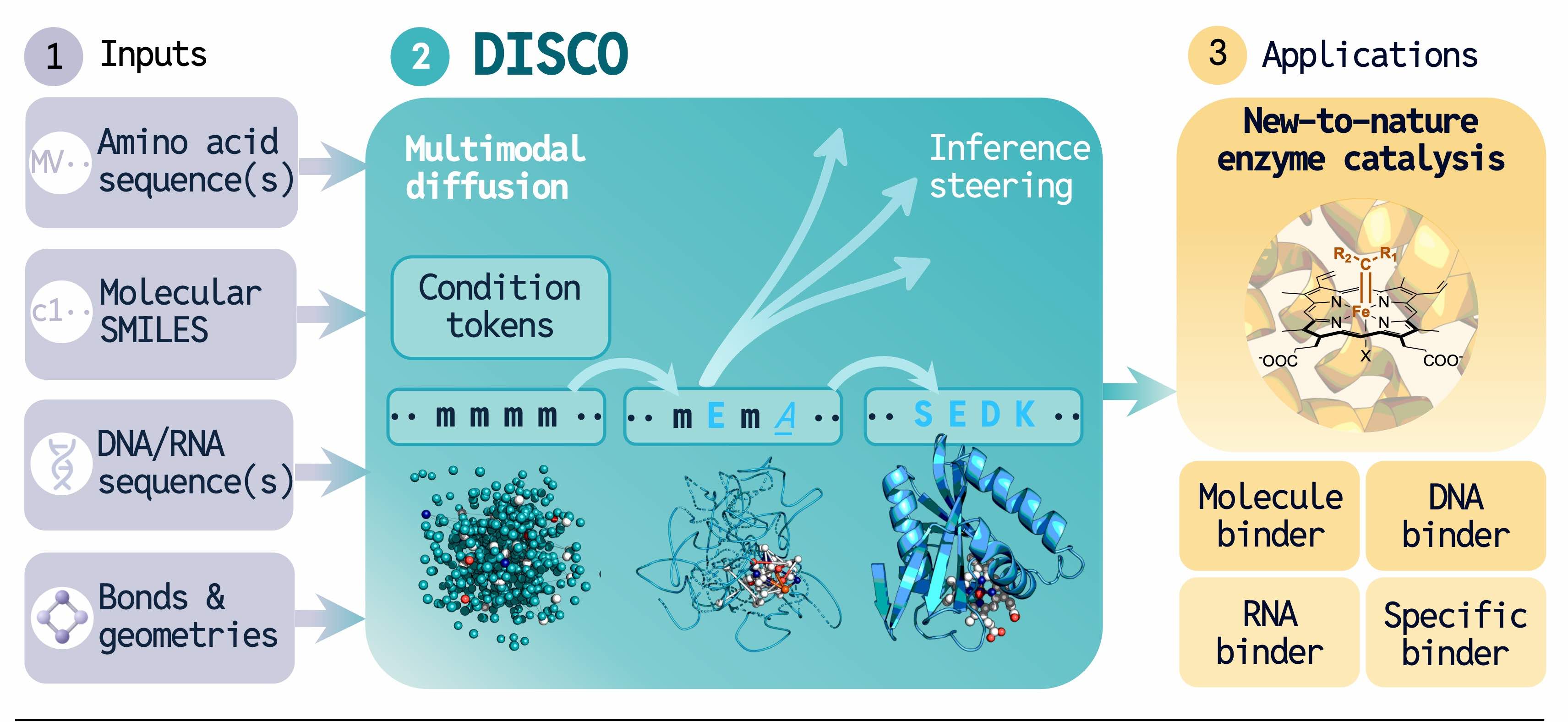
Inference-Time Scaling
Because DISCO operates on both modalities, it unlocks inference-time steering using reward functions defined over both sequence and structure. Rather than generating thousands of candidates and filtering, DISCO uses Feynman-Kac Correctors[11] (FKC) — a principled mathematical framework that tilts the sampling distribution toward proteins with desired properties during generation itself. We derive two novel FKC methods:
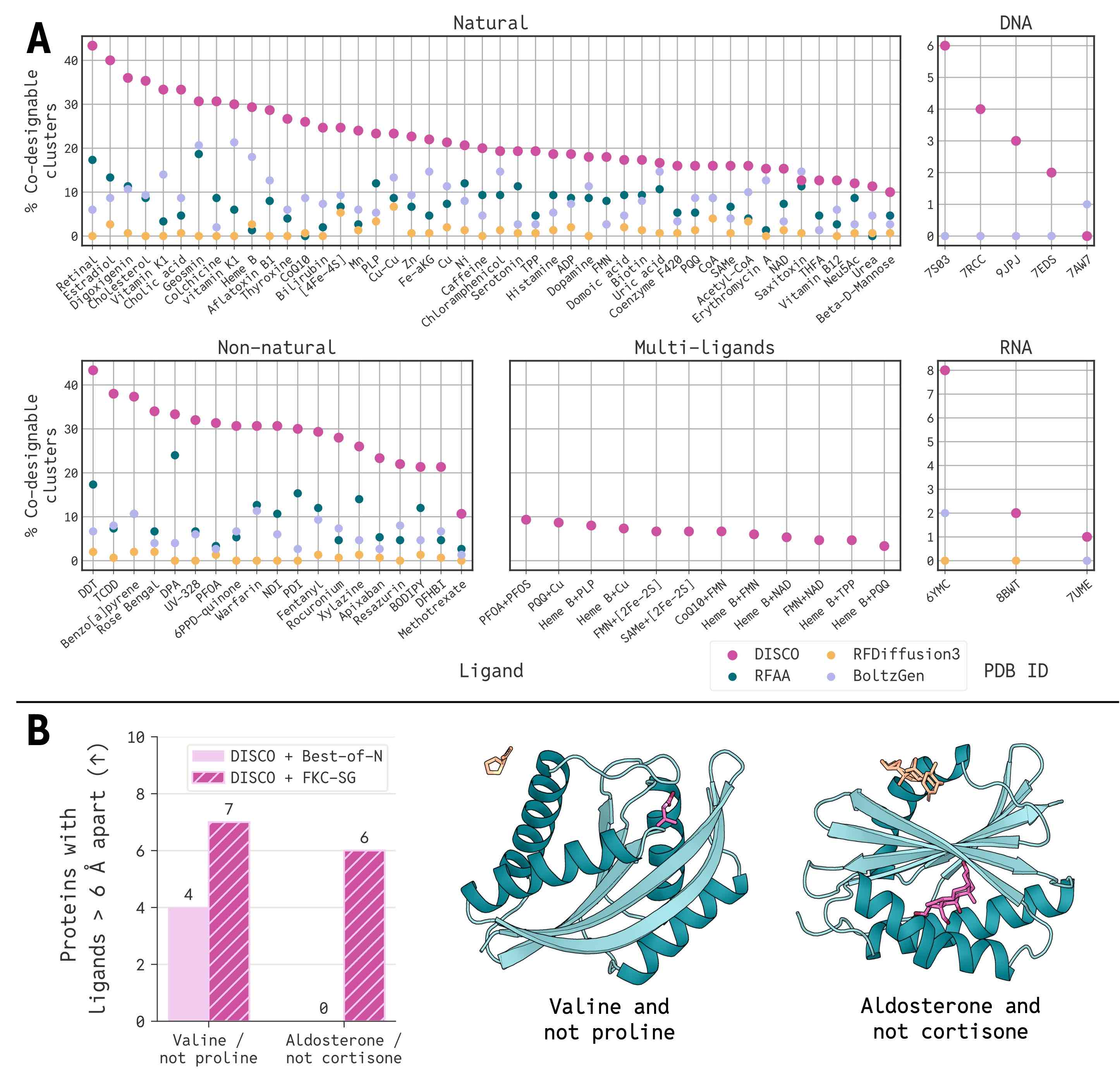
FKC-Multimodal (FKC-MM) steers generation using reward functions defined simultaneously on both discrete sequences and continuous structures, something impossible in decoupled pipelines. When targeting increased disulfide bonds, FKC-MM produces 100-residue proteins with six disulfide bonds, a density found in only the top 0.2% of comparable training proteins. Meanwhile, structure guidance alone does not work.
FKC-Specificity Guidance (FKC-SG) solves a different problem: designing a protein that binds a target molecule while avoiding a structurally similar decoy. By sampling from a tilted distribution that encourages on-target likelihood while penalizing off-target likelihood, FKC-SG generates proteins with high binding-site separation, including cases where best-of-N filtering produces zero hits.